PDF Processor
Inicio
Editar documento
Editar documento: 11. Trombocitopenia (PTI y PTT)
Título
Contenido mejorado
Contenido original
Contenido mejorado por IA (Markdown)
# HEMATOLOGÍA ## TROMBOCITOPENIA ## DEFINICIÓN Se considera trombocitopenia a la disminución del número de plaquetas por debajo de 100,000 plaqueta/mm3. Las disminuciones inferiores a 50,000 plaquetas / mm 3 facilitan el sangrado postraumático y por debajo de 20,000 se facilita el denominado sangrado espontáneo. ## ETIOLOGÍA ## HIPOPRODUCCIÓN DE PLAQUETAS (TROMBOCITOPENIAS CENTRALES) - Disminución del número de megacariocitos: Infiltración de la médula ósea, aplasia, enfermedad de Fanconi, Síndrome TAR (trombopenia y ausencia de radio), trombocitopenia cíclica, rubéola congénita. - Trombopoyesis Ineficaz: Enfermedad de Wiskott-Aldrich, anemias megaloblásticas, Síndromes Mielodisplásicos. ## DISMINUCIÓN DE SUPERVIVENCIA PLAQUETARIA (TROMBOCITOPENIAS PERIFÉRICAS) - Destrucción incrementada de plaquetas (la vida media plaquetaria normal es de alrededor de 10 días): Fármacos, púrpura Trombocitopénica idiopátca, purpura transfusional, púrpura inmunológica secundaria, VIH. - Púpura tromboctiopénica inducida por fármacos: Se produce una destrucción periférica de plaquetas, que ocasiona un incremento en la formación de las mismas mediante un aumento del número de megacariocitos. - Es la trombopenia habitual encontrada en los adultos. - Fármacos causantes: Heparina, etanol, quinidina, difenilhidantoína, sales de oro. | CENTRAL | Disminución producción de plaquetas: Fármacos: Etanol, Tiacidas, Estrógenos, QT | Disminución número de megacariocitos Trombopoyesis inéficaz | | :--: | :--: | :--: | | PERIFÉRICA | - Aumento de la destrucción de plaquetas: - Fármacos, VIH, Auotinmune, Esplenomegalia. - Symbol Consumo: CID, PTT, SHU - Secuestro: Esplenomegalia | | # PÚRPURA TROMBOCITOPÉNICA INMUNE (PTI) ## DEFINICIÓN Se trata de una trombocitopenia originada por fenómenos inmunológicos. Antes llamada Púrpura Trombocitopénica Idiopática. Se puede asociar a Vacuna Hepatits B. - Su etiología es Idiopática en su mayoría (no se asocia a otra patología) - La mayoría se resuelve espontáneamente - No hay tendencia hacia un sexo. ## PATOGENIA Aparición de anticuerpos de tipo IgG sobre la membrana plaquetaria, van dirigidos a antígenos de dicha membrana, como las glucoproteína Ib y lib/Illa. La destrucción de plaquetas ocurre en los macrófagos esplénicos como consecuencia de la presencia de receptores para la fracción constante de IgG en la membrana de dichos macrófagos. Detectar estos Ac Antiplaquetarios tienen sensibilidad 49 - 66% y especificidad 78 - 92 % Deficiencia de anticuerpos contra la metaloproteasa ADAMST13 la cual rompe los grandes multímeros de factor de von Willebrand. ## OLÍNICA Petequias, púrpura, equimosis, epistaxis, gingivorragia, menometrorragia Hemorragia intracraneal y de órganos internos ( <10,000 ) ## PTI AGUDA: - Duración MENOR A 6 MESES. - Suele ser enfermedad infantil, afectando a ambos sexos tras procesos víricos de vía respiratoria alta (80%). - Se recuperan de forma espontánea pero puede haber recurrencia y mortalidad. - No precisan tratamiento. ## PTI CRÓNICA (ENFERMEDAD DE WERLHOF) - Duración >6 MESES. - Suele ser en adultos jóvenes, generalmente mujeres - El 90% no presentan recuperación espontánea y presentan recidivas. - Se deben descartar otras enfermedades como LES - El 65% de los casos está asociada a infección por Hellicobacter pylori 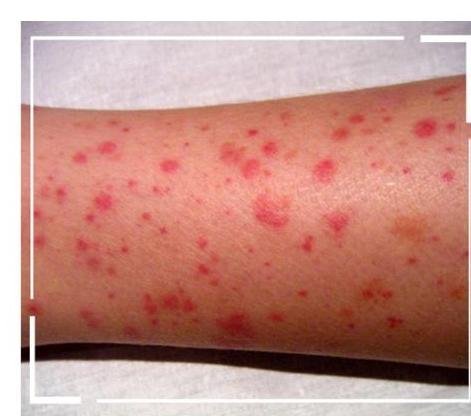 # DIAGNÓSTICO - CLÍNICA apoyada en primera instancia en BH y Frotis de Sangre Periférica (Número de plaquetas y morfología plaquetaria - Demostración de Trombocitopenia de origen inmunológico - Descartar causas posbles de trombocitopenia autoinmune (descartar VIH tiene un cuadro similar), Hepatitis B y C, estudios Inmunológicos (Anticardiolipina, Anticoagulante Lúpico, ANAs) ## TRATAMIENTO ¿A quién se le inicia? - SANGRADO ACTIVO (independientemente del número de plaquetas o <15,000 sin sangrado) - Plaquetas 15,000 - 30,000 (se valorará en función de la edad del paciente y sus comorbilidades). - Plaquetas entre 15,000 y 30,000 y requerirá alguna cirugía. ## 1ra elección: Adminisración de Esteroides - PREDNISONA x 2-4 semanas. - Disminuyen la fagocitosis por los macrófagos y la síntesis de autoanticuerpos. - FALLA DE RESPUESTA - DUPLICAR LA DOSIS. - Si el paciente mantiene cifras plaquetarias <30 \times 10^{\wedge} 9 \rightarrow DANAZOL O IgG anti-D ## 2da elección: ESPLENECTOMÍA - Se elimina el lugar principal de la destrucción plaquetaria y de síntesis de autoanticuerpos. - Si después de la cirugía hay recidiva - PTI Crónica Refractaria: a) Se vuelve a administrar Esteroide b) Fármacos Inmunosupresores - CICLOFOSFAMIDA / AZATIOPRINA / CICLOSPORINA / RITUXIMAB - Indicados si existe falla en los dos tratamientos previos. ## OTROS GAMMAGLOBULINA IV A DOSIS ELEVADAS: bloquea los receptores de la fracción constante de IgG en los macrófagos esplénicos, con lo que la plaqueta no puede unirse al receptor macrofágico. - El tratamiento no es duradero por la vida media del fármaco pero consigue un aumento plaquetario más rápido que ningún otro. Danazol: Produce disminución de la destrucción plaquetaria, al disminuir la expresión de receptores de la fracción constante de la inmunoglobulina G en la mebrana de macrófagos. Plasmaféresis: Elimina los autoanticuerpos. [^0] [^0]: SI EXISTE UN SANGRADO ACTIVO Y TROMBOCITOPENIA SEVERA, SE PREFERIRÁ INICIAR CON TRANSFUSIÓN DE CONCENTRADOS PLAQUETARIOS + GAMMAGLOBULINA IV - esto estabilizará al paciente. UNA VEZ ESTABILIZADO - se podrá iniciar Tx con ESTEROIDES - PREDNISONA. # PÚRPURA TROMBOCITOPÉNICA TROMBÓTICA (PTT) ## DEFINICIÓN También llamado Síndrome de Moschcowitz, es una trombocitopenia de causa desconocida, más frecuente en mujeres de mediana edad, que cursa con una pentada característica: ## TROMBOCITOPENIA CON SANGRADO ## AFECCIÓN NEUROLÓGICA (transitoria y fluctuante) ## FIEBRE DISFUNCIÓN RENAL ANEMIA HEMOLÍTICA MICROANGIOPÁTICA (presencia de esquistocitos en sangre periférica) ## ETIOLOGÍA ## Es desconocida - Antecedente de infección respiratoria alta - Fármacos anovuatorios - Antibióticos - Embarazo (Sx HELLP=Hemolisis, alteración enzimas hepáticas, trombocitopenia, preeclampsia), - LES - Ciclosporina y Mitomicina La patogenia parece tener relación a la existencia de anticuerpos contra la metaloproteasa que degrada el FvW. En su forma hereditaria existe un déficit de la Proteína ADAMTS13, la cual es una metaloproteasa que rompe los grandes multímetros del factor de von Willebrand, desencadenando una exagerada agregación plaquetaria con la formación de trombos plaquetarios dentro de la circulación con el subsecuente consumo de plaquetas y daño mecánico a los eritrocitos y la obstrucción de la microcirculación. ## CLÍNICA - INICIO BRUSCO de la Pentada Clásica: Trombocitopenia con Sangradp + Fiebre + Anemia Hemolítica Micronagiopática + Afección Neurológica + Disfunción Renal. - En etapas tardías se parece a una CIVD. # SÍNDROME HEMOLÍTICO URÉMICO Forma localizada, sin alteración neurológica, con PREDOMINIO RENALE HIPERTENSIÓN ARTERIAL, que aparece fundamentalmente aparece en niños. Existen dos tipos de Síndrome Hemolítico Urémico - TÍPICO: precedido por síntomas gastrointestinales asociados a la SHIGA, toxina producida por la bacteria Shigella disentérica o la E. coli enterohemorrágica (OH 157:H7) productora de VEROTOXINA - ATÍPICO: cuando el SHU no está asociado a la shiga toxina. Solamente el 10% son atípicos. FISIOPATOLOGÍA: Debido a un efecto citotóxico por inhibición de la síntesis protéica que producen las toxinas tipo Shiga y consiguiente ulceración endotelial. ## DIAGNÓSTICO DEL SHU: En pacientes que presenten datos de palidez e ictericia de instalación AGUDA, se deberá solicitar lo sig: - Biometría Hemática con cuenta de reticulocitos - Frotis de sangre periférica - Bilirrubinas - DHL - EGO - Prueba de Coombs ## TRATAMIENTO Si NO SE OTORGA TRATAMIENTO, ES MORTAL EN UN 80 - 90% de los casos. ## 1ra elección: PLASMAFERESIS CON RECAMBIO PLASMÁTICO - Elimina los grandes multímeros del fvW (liberados por las células endoteliales) - Aporta factores inhibidores de la agregación plaquetaria. Alternativa: Esplenectomia / Esteroides / Antiagregantes Plaquetaros / Citoestáticos / Rituximab.
Vista previa
Contenido original (Markdown)
# HEMATOLOGÍA ## TROMBOCITOPENIA ## DEFINICIÓN Se considera trombocitopenia a la disminución del número de plaquetas por debajo de 100,000 plaqueta/mm3. Las disminuciones inferiores a 50,000 plaquetas / mm 3 facilitan el sangrado postraumático y por debajo de 20,000 se facilita el denominado sangrado espontáneo. ## ETIOLOGÍA ## HIPOPRODUCCIÓN DE PLAQUETAS (TROMBOCITOPENIAS CENTRALES) - Disminución del número de megacariocitos: Infiltración de la médula ósea, aplasia, enfermedad de Fanconi, Síndrome TAR (trombopenia y ausencia de radio), trombocitopenia cíclica, rubéola congénita. - Trombopoyesis Ineficaz: Enfermedad de Wiskott-Aldrich, anemias megaloblásticas, Síndromes Mielodisplásicos. ## DISMINUCIÓN DE SUPERVIVENCIA PLAQUETARIA (TROMBOCITOPENIAS PERIFÉRICAS) - Destrucción incrementada de plaquetas (la vida media plaquetaria normal es de alrededor de 10 días): Fármacos, púrpura Trombocitopénica idiopátca, purpura transfusional, púrpura inmunológica secundaria, VIH. - Púpura tromboctiopénica inducida por fármacos: Se produce una destrucción periférica de plaquetas, que ocasiona un incremento en la formación de las mismas mediante un aumento del número de megacariocitos. - Es la trombopenia habitual encontrada en los adultos. - Fármacos causantes: Heparina, etanol, quinidina, difenilhidantoína, sales de oro. | CENTRAL | Disminución producción de plaquetas: Fármacos: Etanol, Tiacidas, Estrógenos, QT | Disminución número de megacariocitos Trombopoyesis inéficaz | | :--: | :--: | :--: | | PERIFÉRICA | - Aumento de la destrucción de plaquetas: - Fármacos, VIH, Auotinmune, Esplenomegalia. - Symbol Consumo: CID, PTT, SHU - Secuestro: Esplenomegalia | | # PÚRPURA TROMBOCITOPÉNICA INMUNE (PTI) ## DEFINICIÓN Se trata de una trombocitopenia originada por fenómenos inmunológicos. Antes llamada Púrpura Trombocitopénica Idiopática. Se puede asociar a Vacuna Hepatits B. - Su etiología es Idiopática en su mayoría (no se asocia a otra patología) - La mayoría se resuelve espontáneamente - No hay tendencia hacia un sexo. ## PATOGENIA Aparición de anticuerpos de tipo IgG sobre la membrana plaquetaria, van dirigidos a antígenos de dicha membrana, como las glucoproteína Ib y lib/Illa. La destrucción de plaquetas ocurre en los macrófagos esplénicos como consecuencia de la presencia de receptores para la fracción constante de IgG en la membrana de dichos macrófagos. Detectar estos Ac Antiplaquetarios tienen sensibilidad 49 - 66% y especificidad 78 - 92 % Deficiencia de anticuerpos contra la metaloproteasa ADAMST13 la cual rompe los grandes multímeros de factor de von Willebrand. ## OLÍNICA Petequias, púrpura, equimosis, epistaxis, gingivorragia, menometrorragia Hemorragia intracraneal y de órganos internos ( <10,000 ) ## PTI AGUDA: - Duración MENOR A 6 MESES. - Suele ser enfermedad infantil, afectando a ambos sexos tras procesos víricos de vía respiratoria alta (80%). - Se recuperan de forma espontánea pero puede haber recurrencia y mortalidad. - No precisan tratamiento. ## PTI CRÓNICA (ENFERMEDAD DE WERLHOF) - Duración >6 MESES. - Suele ser en adultos jóvenes, generalmente mujeres - El 90% no presentan recuperación espontánea y presentan recidivas. - Se deben descartar otras enfermedades como LES - El 65% de los casos está asociada a infección por Hellicobacter pylori  # DIAGNÓSTICO - CLÍNICA apoyada en primera instancia en BH y Frotis de Sangre Periférica (Número de plaquetas y morfología plaquetaria - Demostración de Trombocitopenia de origen inmunológico - Descartar causas posbles de trombocitopenia autoinmune (descartar VIH tiene un cuadro similar), Hepatitis B y C, estudios Inmunológicos (Anticardiolipina, Anticoagulante Lúpico, ANAs) ## TRATAMIENTO ¿A quién se le inicia? - SANGRADO ACTIVO (independientemente del número de plaquetas o <15,000 sin sangrado) - Plaquetas 15,000 - 30,000 (se valorará en función de la edad del paciente y sus comorbilidades). - Plaquetas entre 15,000 y 30,000 y requerirá alguna cirugía. ## 1ra elección: Adminisración de Esteroides - PREDNISONA x 2-4 semanas. - Disminuyen la fagocitosis por los macrófagos y la síntesis de autoanticuerpos. - FALLA DE RESPUESTA - DUPLICAR LA DOSIS. - Si el paciente mantiene cifras plaquetarias <30 \times 10^{\wedge} 9 \rightarrow DANAZOL O IgG anti-D ## 2da elección: ESPLENECTOMÍA - Se elimina el lugar principal de la destrucción plaquetaria y de síntesis de autoanticuerpos. - Si después de la cirugía hay recidiva - PTI Crónica Refractaria: a) Se vuelve a administrar Esteroide b) Fármacos Inmunosupresores - CICLOFOSFAMIDA / AZATIOPRINA / CICLOSPORINA / RITUXIMAB - Indicados si existe falla en los dos tratamientos previos. ## OTROS GAMMAGLOBULINA IV A DOSIS ELEVADAS: bloquea los receptores de la fracción constante de IgG en los macrófagos esplénicos, con lo que la plaqueta no puede unirse al receptor macrofágico. - El tratamiento no es duradero por la vida media del fármaco pero consigue un aumento plaquetario más rápido que ningún otro. Danazol: Produce disminución de la destrucción plaquetaria, al disminuir la expresión de receptores de la fracción constante de la inmunoglobulina G en la mebrana de macrófagos. Plasmaféresis: Elimina los autoanticuerpos. [^0] [^0]: SI EXISTE UN SANGRADO ACTIVO Y TROMBOCITOPENIA SEVERA, SE PREFERIRÁ INICIAR CON TRANSFUSIÓN DE CONCENTRADOS PLAQUETARIOS + GAMMAGLOBULINA IV - esto estabilizará al paciente. UNA VEZ ESTABILIZADO - se podrá iniciar Tx con ESTEROIDES - PREDNISONA. # PÚRPURA TROMBOCITOPÉNICA TROMBÓTICA (PTT) ## DEFINICIÓN También llamado Síndrome de Moschcowitz, es una trombocitopenia de causa desconocida, más frecuente en mujeres de mediana edad, que cursa con una pentada característica: ## TROMBOCITOPENIA CON SANGRADO ## AFECCIÓN NEUROLÓGICA (transitoria y fluctuante) ## FIEBRE DISFUNCIÓN RENAL ANEMIA HEMOLÍTICA MICROANGIOPÁTICA (presencia de esquistocitos en sangre periférica) ## ETIOLOGÍA ## Es desconocida - Antecedente de infección respiratoria alta - Fármacos anovuatorios - Antibióticos - Embarazo (Sx HELLP=Hemolisis, alteración enzimas hepáticas, trombocitopenia, preeclampsia), - LES - Ciclosporina y Mitomicina La patogenia parece tener relación a la existencia de anticuerpos contra la metaloproteasa que degrada el FvW. En su forma hereditaria existe un déficit de la Proteína ADAMTS13, la cual es una metaloproteasa que rompe los grandes multímetros del factor de von Willebrand, desencadenando una exagerada agregación plaquetaria con la formación de trombos plaquetarios dentro de la circulación con el subsecuente consumo de plaquetas y daño mecánico a los eritrocitos y la obstrucción de la microcirculación. ## CLÍNICA - INICIO BRUSCO de la Pentada Clásica: Trombocitopenia con Sangradp + Fiebre + Anemia Hemolítica Micronagiopática + Afección Neurológica + Disfunción Renal. - En etapas tardías se parece a una CIVD. # SÍNDROME HEMOLÍTICO URÉMICO Forma localizada, sin alteración neurológica, con PREDOMINIO RENALE HIPERTENSIÓN ARTERIAL, que aparece fundamentalmente aparece en niños. Existen dos tipos de Síndrome Hemolítico Urémico - TÍPICO: precedido por síntomas gastrointestinales asociados a la SHIGA, toxina producida por la bacteria Shigella disentérica o la E. coli enterohemorrágica (OH 157:H7) productora de VEROTOXINA - ATÍPICO: cuando el SHU no está asociado a la shiga toxina. Solamente el 10% son atípicos. FISIOPATOLOGÍA: Debido a un efecto citotóxico por inhibición de la síntesis protéica que producen las toxinas tipo Shiga y consiguiente ulceración endotelial. ## DIAGNÓSTICO DEL SHU: En pacientes que presenten datos de palidez e ictericia de instalación AGUDA, se deberá solicitar lo sig: - Biometría Hemática con cuenta de reticulocitos - Frotis de sangre periférica - Bilirrubinas - DHL - EGO - Prueba de Coombs ## TRATAMIENTO Si NO SE OTORGA TRATAMIENTO, ES MORTAL EN UN 80 - 90% de los casos. ## 1ra elección: PLASMAFERESIS CON RECAMBIO PLASMÁTICO - Elimina los grandes multímeros del fvW (liberados por las células endoteliales) - Aporta factores inhibidores de la agregación plaquetaria. Alternativa: Esplenectomia / Esteroides / Antiagregantes Plaquetaros / Citoestáticos / Rituximab.
Vista previa
Cancelar
Guardar cambios